Les matériaux à base de carbone ont un énorme potentiel pour construire un avenir durable, mais les scientifiques des matériaux ont besoin d’outils pour analyser correctement leur structure atomique, qui détermine leurs propriétés fonctionnelles. La spectroscopie photoélectronique à rayons X (XPS) est l’un des outils utilisés pour ce faire, mais les résultats XPS peuvent être difficiles à interpréter.
Maintenant, les chercheurs d’Aalto ont développé un outil d’apprentissage automatique pour améliorer les analyses XPS, qu’ils ont mis gratuitement à disposition en tant que Serveur de prédiction XPS.

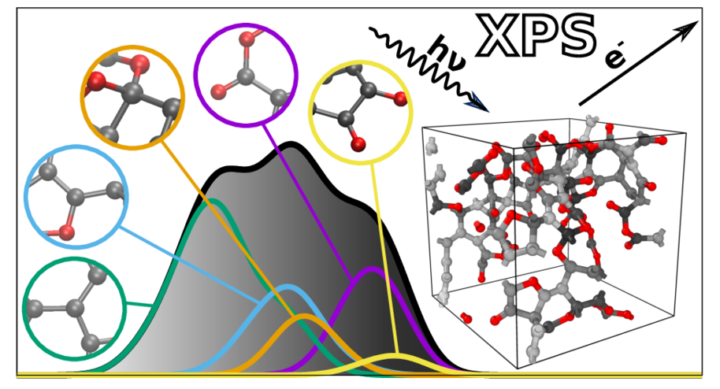
Le nouvel algorithme prédit les spectres XPS de matériaux complexes en fonction des contributions atomiques individuelles. Crédit image : Miguel Caro / Université Aalto
Les spectres XPS sont des graphiques avec une collection de pics qui reflètent l’énergie de liaison des électrons au plus profond des atomes qui composent un matériau. Parce que les énergies de liaison dépendent de l’environnement atomique, elles peuvent être utilisées pour déduire comment les atomes sont connectés dans un matériau ou une molécule particulière. Cependant, cela rend également les spectres XPS difficiles à interpréter, car de nombreux facteurs affectent les énergies de liaison. Les énergies de liaison de différentes caractéristiques atomiques peuvent également se chevaucher, ce qui complique encore l’analyse.
Pour y contribuer, une équipe dirigée par Miguel Caro a développé une méthode de calcul qui peut prédire le spectre d’énergie de liaison d’un matériau sur la base d’un modèle structurel généré par ordinateur. Cela simplifie l’interprétation des données XPS en permettant de faire correspondre les énergies de liaison observées expérimentalement aux prédictions informatiques.
![carte de mise à l'échelle multidimensionnelle basée sur clusterM de la base de données de matériaux cho utilisée dans cette recherche. Crédit image : arXiv : 2112.06551 [cond-mat.mtrl-sci]](https://www.technology.org/texorgwp/wp-content/uploads/2022/07/Screenshot-2022-07-23-at-14-44-10-2112.06551.pdf.png)
carte de mise à l’échelle multidimensionnelle basée sur clusterM du cho
base de données des matériaux utilisés dans cette recherche. Crédit image : arXiv : 2112.06551 [cond-mat.mtrl-sci]
L’idée elle-même n’est pas nouvelle, mais le problème a été la difficulté informatique de calculer avec précision le spectre XPS d’un matériau. L’équipe de Caro a résolu ce problème en utilisant l’apprentissage automatique. L’astuce consistait à former un algorithme informatique peu coûteux pour prédire le résultat d’une méthode de référence coûteuse en calcul basée sur une combinaison efficace de données de mécanique quantique coûteuses et bon marché en calcul.
La méthode la moins coûteuse en termes de calcul, DFT, ne correspond pas très précisément aux résultats expérimentaux. La méthode la plus précise, GW, prend trop de temps à calculer lorsqu’une molécule a de nombreux atomes. «Nous avons décidé de construire un modèle de base qui utilise d’abondantes données DFT, puis de l’affiner avec des données GW rares et précieuses. Et ça a marché !’ dit Caro.
L’algorithme résultant peut prédire le spectre de tout matériau désordonné composé de carbone, d’hydrogène et d’oxygène. «Les spectres prédits sont remarquablement proches de ceux obtenus expérimentalement. Cela ouvre la porte à une meilleure intégration entre la caractérisation expérimentale et informatique des matériaux», déclare Caro. Ensuite, l’équipe prévoit d’étendre sa technique pour inclure une gamme plus large de matériaux et d’autres types de spectroscopie.
La source: Université Aalto





