La sclérose latérale amyotrophique (SLA) est une maladie dégénérative rapidement progressive et mortelle affectant les cellules nerveuses du cerveau et de la moelle épinière responsables du contrôle des mouvements musculaires volontaires. La SLA « sporadique » ou non héréditaire représente environ 90 % des cas et 10 % des cas sont dus à des mutations génétiques connues.
En étudiant les neurones cultivés en laboratoire dérivés de la peau ou des cellules sanguines de 10 témoins normaux, huit avec une mutation provoquant la SLA et 17 avec une SLA non héréditaire, les chercheurs ont trouvé un point de départ possible pour le dysfonctionnement qui cause la maladie. L’étude, publiée dans Science Médecine translationnelle a été financé en partie par le National Institute for Neurological Disorders and Stroke (NINDS), qui fait partie des National Institutes of Health.

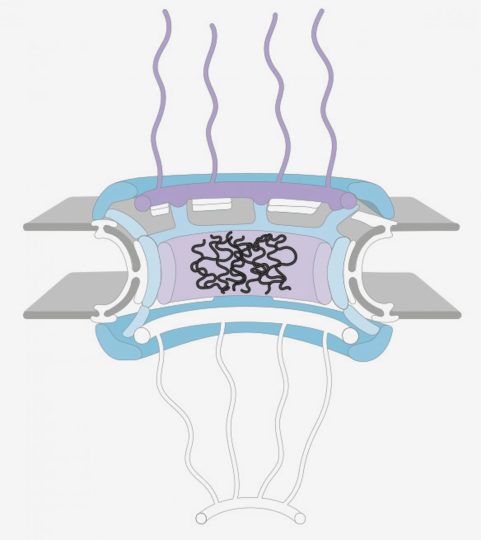
Lorsque CHMP7 s’accumule dans le noyau, certaines protéines disparaissent des pores nucléaires (encadrées en blanc). Cela provoque la rupture des pores, entraînant des effets en aval pouvant provoquer la SLA. Crédit Imahge: Rothstein Lab
À l’aide d’une bibliothèque de cellules dérivées de patients atteints de SLA, l’équipe de recherche dirigée par Jeffrey Rothstein, MD, Ph.D., à la Johns Hopkins University School of Medicine, Baltimore, a développé des neurones dérivés de cellules souches pluripotentes induites (iPSC) à partir des patients cellules cultivées pour découvrir un défaut commun, que la cellule provienne de personnes atteintes de SLA héréditaire ou non. Ils rapportent que dans les cellules nerveuses de la SLA, il y a une accumulation d’une protéine appelée CHMP7 dans le noyau des cellules nerveuses en culture ainsi que dans les échantillons de SLA de la région du cerveau qui contrôle le mouvement. Les traitements qui diminuent la quantité de CHMP7 dans les cellules cultivées ont empêché une série d’anomalies caractéristiques de la SLA.
« Il existe un intérêt considérable pour l’identification de nouvelles cibles thérapeutiques pour la SLA, en particulier pour la forme sporadique de la maladie », a déclaré Amélie Gubitz, Ph.D., directrice de programme, NINDS. « Les stratégies de ciblage génétique comme celle présentée ici nous permettent désormais de passer directement de la découverte biologique au développement de thérapies. »
Cette étude s’appuie sur un article antérieur du laboratoire Rothstein qui a examiné la cause génétique la plus courante de la SLA, une mutation du gène C9orf72 (également appelée « mutation C9 »). Là, ils ont montré que la mutation C9 produisait des défauts dans une structure appelée pore nucléaire qui est responsable du déplacement des protéines et d’autres molécules dans et hors du noyau des cellules. Plus précisément, ils ont découvert que certaines protéines étaient absentes du pore, provoquant un effet de type domino dans lequel le pore entier se brise.
« Nous savions grâce à nos travaux précédents que la mutation C9 produisait des défauts dans le pore nucléaire, mais nous ne savions pas pourquoi », a déclaré le Dr Rothstein. « Ici, nous avons entrepris de répondre à la question de ce qui se passait en amont des défauts des pores en étudiant les neurones dérivés des cellules de patients atteints de SLA. »
Plus précisément, les chercheurs ont examiné les cellules nerveuses cultivées à partir de cellules souches pluripotentes induites (iPSC), qui sont un type de cellules souches pouvant être créées à partir d’échantillons de peau ou de sang d’une personne. Ces cellules se comportent de manière très similaire aux autres cellules souches en ce sens qu’elles peuvent être transformées en de nombreux types de cellules différents en laboratoire, y compris des cellules nerveuses. En travaillant avec Answer ALS, un effort national de données biologiques sur la SLA et d’iPSC dirigé par Rothstein, les chercheurs ont pu accéder aux iPSC dérivés de patients SLA familiaux et sporadiques.
« L’un des grands avantages des iPSC est que vous pouvez examiner différents moments de la même manière que vous étudieriez des modèles animaux à différents âges », a déclaré le Dr Rothstein. « Nous savions à quel moment les pores nucléaires ont commencé à se dégrader, et nous avons pu étudier les neurones à des époques antérieures pour voir quelle pouvait en être la cause. »
Ce qu’ils ont découvert, c’est que l’accumulation de CHMP7 dans le noyau s’est produite au moins une semaine avant le développement d’anomalies des pores nucléaires. Normalement, CHMP7 est rapidement éliminé une fois qu’il pénètre dans le noyau, mais dans les neurones C9 et sporadiques dérivés des iPSC de la SLA, l’accumulation a persisté. Si un médicament oligonucléotidique antisens, qui empêche les cellules de fabriquer des protéines spécifiques, a été utilisé pour diminuer la quantité de CHMP7 dans les neurones de la SLA, le pore ne s’est jamais dégradé. Enfin, si une forme mutée de CHMP7 qui ne peut pas être retirée du noyau était ajoutée à des neurones sains, le pore se dégradait un peu comme ce qui a été observé dans les neurones de la SLA, suggérant que la présence de CHMP7 dans les noyaux des neurones pourrait être un événement clé dans le développement de la maladie.
Une anomalie commune à toutes les formes de SLA est la mauvaise localisation d’une autre protéine, la TDP-43. Normalement présent dans le noyau, le TDP-43 s’infiltre dans le cytoplasme environnant dans la SLA où il s’agglutine en agrégats, entraînant une perte de fonctions dans divers types d’ARN, qui sont essentiels à la traduction de certains gènes en protéines. Finalement, cela se voit également dans les neurones dérivés d’iPSC provenant à la fois de patients atteints de SLA C9 et sporadiques. Suite au traitement avec les oligonucléotides antisens pour CHMP7, la mauvaise localisation de TDP-43 n’a plus été observée et les défauts d’ARN ont tous été corrigés.
« Ces résultats nous permettent de mettre ces anomalies dans l’ordre, où l’accumulation de CHMP7 dans le noyau entraîne une lésion des pores nucléaires, suivie d’une mauvaise localisation du TDP-43 et, finalement, de la mort cellulaire », a déclaré le Dr Rothstein. « Ceci ne se limite pas à la mutation C9 ; c’est également une voie fondamentale dans la SLA sporadique qui peut être traitée avec des oligonucléotides antisens pour CHMP7.
Le laboratoire du Dr Rothstein cherche actuellement à savoir si l’oligonucléotide antisens pourrait être développé en un traitement pour les patients atteints de SLA C9 et sporadique. Ils continuent également à étudier l’accumulation initiale de CHMP7 pour déterminer les causes de la mauvaise localisation dans le noyau.
La source: NIH





